Sanjar Hudaiberdiev
I am a Computational Biologist passionate about applying ML/AI to biological problems.
- 13 years of work experience in computational biology (10 postgrad).
- 6 years of machine learning exerience.
- Expertise in Python, R, SQL, and modern data science workflows.
- Experience in Software Development (OOP, functional programming, Git, Unit testing).
- A deep understanding of modern genetics and genomics methods.
- Skilled writer and communicator.
- Research leadership roles, editorial experience, and project management.
selected publications
-
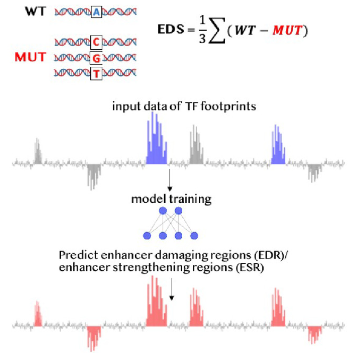 Modeling islet enhancers using deep learning identifies candidate causal variants at loci associated with T2D and glycemic traitsSanjarbek Hudaiberdiev, D Leland Taylor , Wei Song , and 8 more authorsProceedings of the National Academy of Sciences, 2023
Modeling islet enhancers using deep learning identifies candidate causal variants at loci associated with T2D and glycemic traitsSanjarbek Hudaiberdiev, D Leland Taylor , Wei Song , and 8 more authorsProceedings of the National Academy of Sciences, 2023Genetic association studies have identified hundreds of independent signals associated with type 2 diabetes (T2D) and related traits. Despite these successes, the identification of specific causal variants underlying a genetic association signal remains challenging. In this study, we describe a deep learning (DL) method to analyze the impact of sequence variants on enhancers. Focusing on pancreatic islets, a T2D relevant tissue, we show that our model learns islet-specific transcription factor (TF) regulatory patterns and can be used to prioritize candidate causal variants. At 101 genetic signals associated with T2D and related glycemic traits where multiple variants occur in linkage disequilibrium, our method nominates a single causal variant for each association signal, including three variants previously shown to alter reporter activity in islet-relevant cell types. For another signal associated with blood glucose levels, we biochemically test all candidate causal variants from statistical fine-mapping using a pancreatic islet beta cell line and show biochemical evidence of allelic effects on TF binding for the model-prioritized variant. To aid in future research, we publicly distribute our model and islet enhancer perturbation scores across 67 million genetic variants. We anticipate that DL methods like the one presented in this study will enhance the prioritization of candidate causal variants for functional studies.
@article{hudaiberdiev2023modeling, title = {Modeling islet enhancers using deep learning identifies candidate causal variants at loci associated with T2D and glycemic traits}, author = {Hudaiberdiev, Sanjarbek and Taylor, D Leland and Song, Wei and Narisu, Narisu and Bhuiyan, Redwan M and Taylor, Henry J and Tang, Xuming and Yan, Tingfen and Swift, Amy J and Bonnycastle, Lori L and others}, journal = {Proceedings of the National Academy of Sciences}, volume = {120}, number = {35}, pages = {e2206612120}, year = {2023}, google_scholar_id = {Se3iqnhoufwC}, url = {https://www.pnas.org/doi/full/10.1073/pnas.2206612120?doi=10.1073/pnas.2206612120} } -
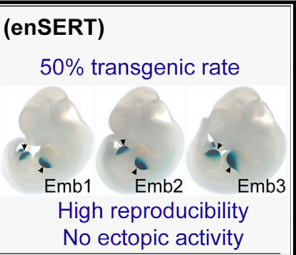 Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variantsEvgeny Z Kvon , Yiwen Zhu , Guy Kelman , and 8 more authorsCell, 2020
Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variantsEvgeny Z Kvon , Yiwen Zhu , Guy Kelman , and 8 more authorsCell, 2020Establishing causal links between non-coding variants and human phenotypes is an increasing challenge. Here, we introduce a high-throughput mouse reporter assay for assessing the pathogenic potential of human enhancer variants in vivo and examine nearly a thousand variants in an enhancer repeatedly linked to polydactyly. We show that 71% of all rare non-coding variants previously proposed as causal lead to reporter gene expression in a pattern consistent with their pathogenic role. Variants observed to alter enhancer activity were further confirmed to cause polydactyly in knockin mice. We also used combinatorial and single-nucleotide mutagenesis to evaluate the in vivo impact of mutations affecting all positions of the enhancer and identified additional functional substitutions, including potentially pathogenic variants hitherto not observed in humans. Our results uncover the functional consequences of hundreds of mutations in a phenotype-associated enhancer and establish a widely applicable strategy for systematic in vivo evaluation of human enhancer variants.
@article{kvon2020comprehensive, title = {Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants}, author = {Kvon, Evgeny Z and Zhu, Yiwen and Kelman, Guy and Novak, Catherine S and Plajzer-Frick, Ingrid and Kato, Momoe and Garvin, Tyler H and Pham, Quan and Harrington, Anne N and Hunter, Riana D and others}, journal = {Cell}, volume = {180}, number = {6}, pages = {1262--1271}, year = {2020}, publisher = {Elsevier}, google_scholar_id = {WF5omc3nYNoC}, url = {https://www.sciencedirect.com/science/article/pii/S0092867420302087} } -
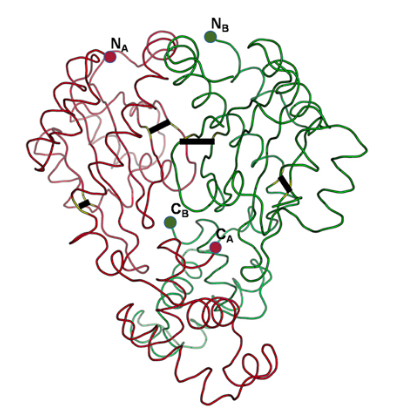 Census of solo LuxR genes in prokaryotic genomesSanjarbek Hudaiberdiev, Kumari S Choudhary , Roberto Vera Alvarez , and 4 more authorsFrontiers in cellular and infection microbiology, 2015
Census of solo LuxR genes in prokaryotic genomesSanjarbek Hudaiberdiev, Kumari S Choudhary , Roberto Vera Alvarez , and 4 more authorsFrontiers in cellular and infection microbiology, 2015luxR genes encode transcriptional regulators that control acyl homoserine lactone-based quorum sensing (AHL QS) in Gram negative bacteria. On the bacterial chromosome, luxR genes are usually found next or near to a luxI gene encoding the AHL signal synthase. Recently, a number of luxR genes were described that have no luxI genes in their vicinity on the chromosome. These so-called solo luxR genes may either respond to internal AHL signals produced by a non-adjacent luxI in the chromosome, or can respond to exogenous signals. Here we present a survey of solo luxR genes found in complete and draft bacterial genomes in the NCBI databases using HMMs. We found that 2698 of the 3550 luxR genes found are solos, which is an unexpectedly high number even if some of the hits may be false positives. We also found that solo LuxR sequences form distinct clusters that are different from the clusters of LuxR sequences that are part of the known luxR-luxI topological arrangements. We also found a number of cases that we termed twin luxR topologies, in which two adjacent luxR genes were in tandem or divergent orientation. Many of the luxR solo clusters were devoid of the sequence motifs characteristic of AHL binding LuxR proteins so there is room to speculate that the solos may be involved in sensing hitherto unknown signals. It was noted that only some of the LuxR clades are rich in conserved cysteine residues. Molecular modeling suggests that some of the cysteines may be involved in disulfide formation, which makes us speculate that some LuxR proteins, including some of the solos may be involved in redox regulation.
@article{hudaiberdiev2015census, title = {Census of solo LuxR genes in prokaryotic genomes}, author = {Hudaiberdiev, Sanjarbek and Choudhary, Kumari S and Vera Alvarez, Roberto and Gelencs{\'e}r, Zsolt and Ligeti, Bal{\'a}zs and Lamba, Doriano and Pongor, S{\'a}ndor}, journal = {Frontiers in cellular and infection microbiology}, volume = {5}, pages = {20}, year = {2015}, publisher = {Frontiers Media SA}, google_scholar_id = {2osOgNQ5qMEC}, url = {https://www.frontiersin.org/articles/10.3389/fcimb.2015.00020/full} } -
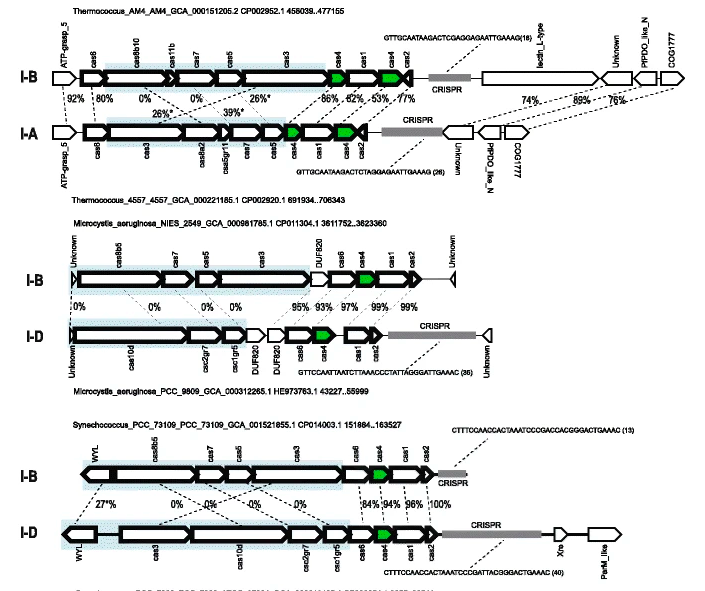 Phylogenomics of Cas4 family nucleasesSanjarbek Hudaiberdiev, Sergey Shmakov , Yuri I Wolf , and 3 more authorsBMC evolutionary biology, 2017
Phylogenomics of Cas4 family nucleasesSanjarbek Hudaiberdiev, Sergey Shmakov , Yuri I Wolf , and 3 more authorsBMC evolutionary biology, 2017The Cas4 family endonuclease is a component of the adaptation module in many variants of CRISPR-Cas adaptive immunity systems. Unlike most of the other Cas proteins, Cas4 is often encoded outside CRISPR-cas loci (solo-Cas4) and is also found in mobile genetic elements (MGE-Cas4). As part of our ongoing investigation of CRISPR-Cas evolution, we explored the phylogenomics of the Cas4 family. About 90% of the archaeal genomes encode Cas4 compared to only about 20% of the bacterial genomes. Many archaea encode both the CRISPR-associated form (CAS-Cas4) and solo-Cas4, whereas in bacteria, this combination is extremely rare. The solo-cas4 genes are over-represented in environmental bacteria and archaea with small genomes that typically lack CRISPR-Cas, suggesting that Cas4 could perform uncharacterized defense or repair functions in these microbes. Phylogenomic analysis indicates that both the CRISPR-associated cas4 genes are often transferred horizontally but almost exclusively, as part of the adaptation module. The evolutionary integrity of the adaptation module sharply contrasts the rampant shuffling of CRISPR-cas modules whereby a given variant of the adaptation module can combine with virtually any effector module. The solo-cas4 genes evolve primarily via vertical inheritance and are subject only to occasional horizontal transfer. The selection pressure on cas4 genes does not substantially differ between CAS-Cas4 and solo-cas4, and is close to the genomic median. Thus, cas4 genes, similarly to cas1 and cas2, evolve similarly to ‘regular’ microbial genes involved in various cellular functions, showing no evidence of direct involvement in virus-host arms races. A notable feature of the Cas4 family evolution is the frequent recruitment of cas4 genes by various mobile genetic elements (MGE), particularly, archaeal viruses. The functions of Cas4 in these elements are unknown and potentially might involve anti-defense roles. Unlike most of the other Cas proteins, Cas4 family members are as often encoded by stand-alone genes as they are incorporated in CRISPR-Cas systems. In addition, cas4 genes were repeatedly recruited by MGE, perhaps, for anti-defense functions. Experimental characterization of the solo and MGE-encoded Cas4 nucleases is expected to reveal currently uncharacterized defense and anti-defense systems and their interactions with CRISPR-Cas systems.
@article{hudaiberdiev2017phylogenomics, title = {Phylogenomics of Cas4 family nucleases}, author = {Hudaiberdiev, Sanjarbek and Shmakov, Sergey and Wolf, Yuri I and Terns, Michael P and Makarova, Kira S and Koonin, Eugene V}, journal = {BMC evolutionary biology}, volume = {17}, number = {1}, pages = {1--14}, year = {2017}, publisher = {BioMed Central}, google_scholar_id = {Y0pCki6q_DkC}, url = {https://bmcecolevol.biomedcentral.com/articles/10.1186/s12862-017-1081-1} }



